
Since I'm on-route to a Human Microbiome Project meeting (uncharacteristically, it's being held in a climate-friendly location--Houston; last year, it was held in Boston. In January.), reviewing this paper about the GEBA project, the Genomic Encyclopedia of Bacteria and Archaea, seemed appropriate. Sequencing bacterial genomes not only tells us a lot about the biology of the organisms sequenced, including their function, potential ecology, and evolution, but it also has a far more pragmatic use too.
As we try to understand microbial communities using DNA sequencing, including those microbes living on us (the human microbiome), we are increasingly moving towards whole genome shotgun methods: the DNA is extracted from a sample of interest and sequenced in its entirety. What this yields is a lot of short 'reads'--sequences of DNA that typically don't cover an entire gene, only some of which can be assembled into larger genomic fragments*.
Nonetheless, we can still, in many cases, identify what the gene is (and does) and what organism it came from. This task is much easier if a gene similar to the one you're trying to identify has already been discovered in a genome. In other words, over the next five to ten years, if we want to understand microbial ecology as well as human health and disease, we need more genomes. However, we not only need more genomes, but 'better' ones. What do I mean by better? Our current collection of genomes is biased towards those organisms of medical importance and towards organisms that are easy to grow (or possible to grow).
So back to GEBA. The GEBA project is attempting to fill in the genomic gaps--that is, sequence bacteria from groups that are underrepresented or not represented at all. Below, mapped onto a phylogeny (how bacterial species are related to each other) in red, are the GEBA-sequenced genomes:
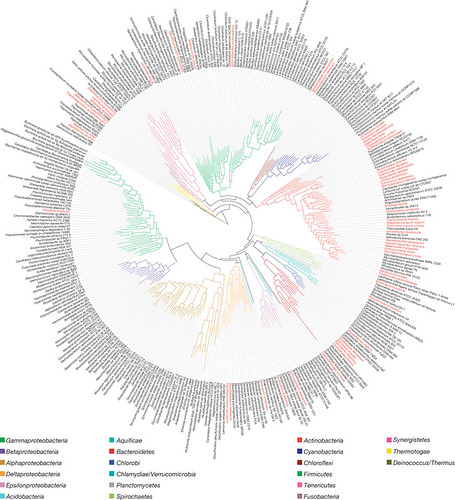
(click to embiggen)
{kind=link}
So what does sequencing phylogenetically undersampled genomes mean for discovering new genes? Well, we get far genetic bang for our genomic buck--we discover many more novel 'gene families' (gene classes) per genome:
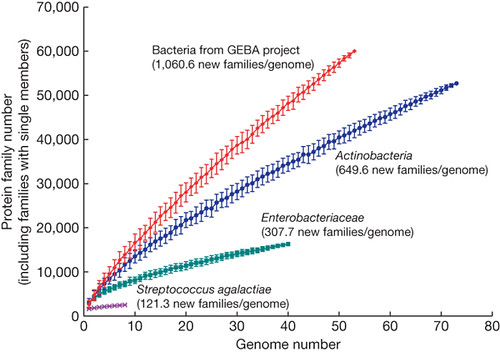
As we move from sampling within a species (the purple line), to within a family (green) to within a phylum (blue), the number of new gene families increases (as one would expect--more diverse organism, more novel diversity). But GEBA blows all of this away. This will be very useful for genomics.
Since you made it this far, we end with the musical portion of our program (although tastes might differ...):
*One advantage of WGS over 16S rDNA sequencing is that WGS doesn't have PCR biases. On the other hand, the informatics are much harder.
Related post: Byte Size Biology has a good review of the paper and an interesting interview with one of the authors, Jonathan Eisen.
OK - that musical portion, well, it rocks. Maybe I will include it at the end of all my talks on GEBA