
A lot of readers (well, a couple, anyway) have been asking me about the recent article by Peter Duesberg in the most recent issue of Scientific American entitled Chromosomal Chaos and Cancer. I suppose it's because I'm not only a cancer surgeon (which in and of itself is not enough to qualify me to comment on this topic) but rather because I'm also a cancer researcher and a molecular biologist (which, I submit, does make me qualified to comment on this topic). Peter Duesberg, as you may know, is the controversial scientist who is perhaps the foremost advocate of the discredited hypothesis that HIV does not cause AIDS. I had been tempted to comment on Dueseberg's hypothesis based on the orgasmic reaction of Duesberg's sycophants in the HIV/AIDS "dissident" community to the recent publication of an article on it by Duesberg in Scientific American. One such booster, even went so far as to say:
The one that first comes to mind as particularly relevant to Peter and AIDS is that it does seem impossible that a man who might just be correct concerning something as complicated as the genetic basis of malignancy could be so totally wrong about something as straightforward as whether HIV kills T-cells.
This is perhaps the dumbest thing I've heard said about evaluating Duesberg's aneuploidy hypothesis of cancer. To demonstrate why, let me recast the question a bit to: "The one that first comes to mind as particularly relevant to Peter and AIDS is that it does seem impossible that a man who is clearly so utterly wrong concerning something as now scientifically straightforward as whether AIDS is caused by HIV could actually be correct about something as complex as the genetic basis of malignancy."
HIV/AIDS denialists would be screaming bloody murder about such a question were I to pose it as anything other than a rhetorical device, and they would have a point to some extent. Yet they think nothing conflating the scientific validity of Duesberg's ideas concerning cancer, which might indeed be partially or mostly correct, with his discredited hypothesis that HIV does not cause AIDS, implying that because he might be correct about cancer implies that he is correct about AIDS. It doesn't. Sorry, but the two issues are at best peripherally and weakly related and at most not related at all. That is why I do not plan on discussing why Duesberg's ideas regarding HIV are wrong, even though the results of such HIV/AIDS denialism leads to quackery and has serious real-world consequences for real people. Tara and Nick have done far more than my minor efforts to critically examine such wingnuttery. Instead, I decided simply to ask the question about Duesberg's chromosomal chaos hypothesis as the cause of cancer and ask: Is there any "there" there? It's a question I've asked myself before but never written about, and this seemed like an opportune time to discuss the issue.
The first thing that struck me about the Scientific American article is that it looked very much like a popular version of two very similar recent review/opinion articles that Duesberg published in 2005 and 2006. I'm mainly going to discuss the Scientific American article because it's basically the same message in a form more palatable to the educated lay reader. But, before I begin, I'd like to point out a couple of things. First, the concept that chromosomal abnormalities cause cancer dates back at least to 1914, when the German zoologist Theodor Boveri, based on studies of sea urchin development, first suggested it. Indeed, this featured prominently as a milestone in cancer research in a display in at the recent 100th anniversary meeting of the American Association for Cancer Research. Thus, the basis of Duesberg's idea is quite old. Indeed, the concept that chromosomal derangements caused cancer predominated for 40-50 years, until the solution to the structure of DNA, the elucidation of the genetic code, and study of genetics led to an emphasis on genetic causes of cancer. Combined with the observation that tumor cells are genetically unstable, leading to many mutations, the genetic hypothesis led to the discovery of oncogenes and tumor suppressors. Now, potential chromosomal causes are again being looked at, and for whatever part Duesberg's advocacy had in spurring this he is to be acknowledged, even if his boosters do have an annoying tendency to make it sound as though scientists would have zero interest in studying chromosomal causes of cancer were it not for Duesberg, which, given the attention shown to this topic at recent meetings that I've attended, is ridiculous.
It has been known for many many decades that most cancer cells are aneuploid. This means that, instead of having the correct number of chromosomes (in the human, 46 chromosomes, two matched sets of 22, plus the sex chromosomes, either XX or XY). In some diseases, such as Down syndrome (known as trisomy-21), there are either missing or extra chromosomes. In the case of Down syndrome, there is a third copy of chromosome 21. Such abnormal chromosomal numbers come about when, during meisosis (cell division that produce germ cells), the newly copied chromosomes don't segregate properly to the two daughter cells, one to each. Instead, both go to one or the other cell. In cancer cells, the situation is much worse; such missegregation during mitosis can lead to aneuploidy that is much more severe, to the point where some cancer cells can have 70 chromosomes or more. Because certain genetic mutations, for example in DNA damage repair genes, can lead to chromosomal instability that can in turn lead to aneuploidy, the basic argument has been over the relative importance of the roles of aneuploidy and the accumulation of genetic mutations as leading to cancer. On the one extreme, there is the argument that aneuploidy is the primary cause of cancer, causing the accumulation of genetic mutations through breaks in chromosomes. On the other extreme is the argument that aneuploidy is a consequence, not a cause, of cancer. Duesberg, as you may guess, takes the extreme version of the former view. These days, most other scientists studying this question tend to consider both important to varying degrees in the development of cancer, the present pressing scientific question being: Which causes which and how? It's very much a chicken-or-the-egg problem. Does mutation lead to aneuploidy or aneuploidy lead to large numbers of genetic derangements that lead to cancer? Or are both aneuploidy and mutation responsible in differing proportions depending on the cancer?
Unfortunately, although he describes how tumors evolve under the selective pressures of the organs in which they arise, acquiring the ability to proliferate, evade apoptosis, become insensitive to normal growth arrest signals, and metastasize, Duesberg seems unable to restrain himself from overselling his case and lapsing into straw men arguments about rival hypotheses. He's very frustrating that way. The concept that aneuploidy may play a major and, in some cases, primary role in carcinogenesis is a legitimate scientific idea with scientific evidence to support it. It doesn't need to be defended or argued using such bad arguments, but Duesberg can't seem to help himself. The problem is, Duesberg's thinking is black-and-white, all-or-nothing. He can't seem to fathom the concept that both aneuploidy and genetic mutations might feed upon each other. In addition, he also almost totally neglects other evidence implicating other causes or important factors in the progression of cancer, such as cancer stem cells, tumor angiogenesis, or even the metabolic hypothesis ( i.e., the Warburg effect), which, like the chromosomal hypothesis, is also enjoying a resurgence. Cancer is a complex set of diseases, likely with multiple causes contributing to the development of different cancers in different proportions, which is why my skeptical antennae start twitching whenever I hear someone like Duesberg (or anyone else, for that matter) postulate in essence a single cause for all cancer.
Let's look at what Duesberg argues. In essence, he argues that aneuploidy comes first and is the prime inciting event that starts the cascade of genetic changes that lead to malignancy. DNA is damaged, either through mutagens or other causes, and then, through what becomes a self-catalyzing process, aneuploidy leads to progressive chromosomal alterations that lead to increasingly widespread genetic alterations in a process that feeds on itself, leading to chromosomal instability and cancer. Indeed, Duesberg postulates that carcinogens work as "aneuploidogens" rather than as mutagens.
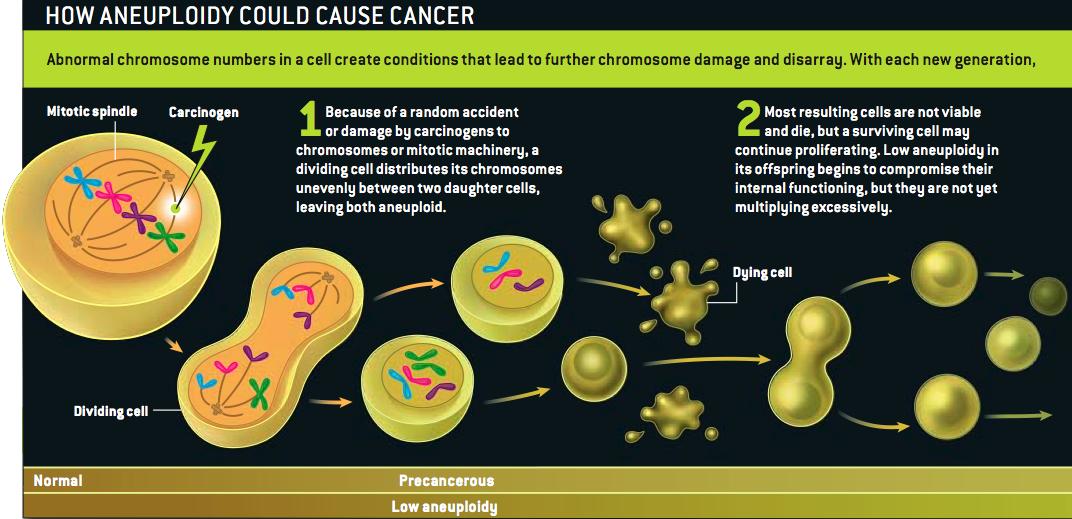
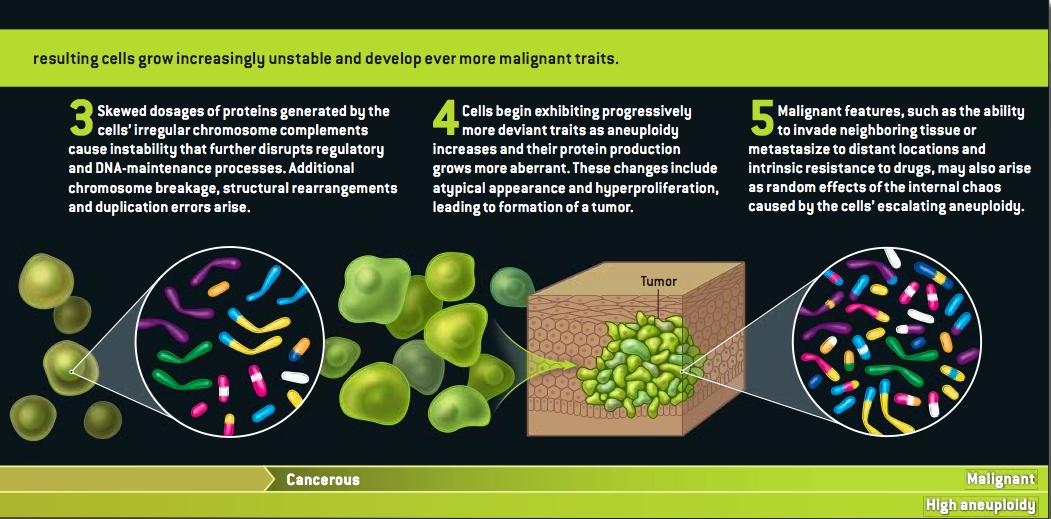
Grrlscientist has posted a concise summary of Duesberg's arguments here, and Hank Barnes has a copy here. (I'm guessing that Duesberg or Harvey Bialy gave him the PDF.)
So, here we have a scientific hypothesis with a moderate degree of plausibility based on what we know. What arguments can Duesberg marshal in its favor? Disappointingly, most of his arguments leave much to be desired. Let's start with Duesberg's first argument, which is so bad that it needs to be quoted extensively to be appreciated:
Cancer risk grows with age. Lamentably common, cancer afflicts about one in three people at some point in their lives, but mostly after the age of 50, which is when chances for malignancy soar. Thus, cancer is, by and large, a disease of old age. The gene mutation theory of cancer's origins, however, predicts that the disease should be quite common in newborns. If, as that hypothesis holds, about half a dozen mutations to critical genes were necessary to ignite malignancy, certainly some of those mutations would accumulate like SNPs over the course of generations in the genomes of many individuals. A baby could thus inherit three of six hypothetical colon cancer mutations from her mother, for example, and two from her father and be at extremely high risk of cancer from picking up the missing sixth mutation in any one of her billions of colon cells. Some babies might even be born with colon cancer from inheriting all six hypothetical colon cancer mutations from their parents. But colon cancer is never seen in children. Indeed, even laboratory mice intentionally engineered to carry an assortment of ostensibly carcinogenic mutations from birth can live and propagate happily, with no higher risk of developing tumors than normal lab mice.
[...]
Interestingly, among the rare exceptions to cancer's age bias are children with congenital aneuploidy, as in Down syndrome, or with inherited chromosome instability syndromes, such as the disease known as mosaic variegated aneuploidy (MVA), which also causes severe mental retardation. Defects of mitotic spindle assembly in the cells of children with MVA produce random aneuploidies throughout their bodies, and nearly one third develop leukemia or unusual solid cancers.
Being born aneuploid, or prone to aneuploidy, clearly accelerates processes that lead to cancer. Indeed, the inherent instability of aneuploid cells would explain why most aneuploid embryos, as Boveri observed 100 years ago, would not be viable at all and thus why newborns are cancer-free and cancer is not heritable.
.
This argument is a strawman and neglects other factors, to boot. For one thing, contrary to what Duesberg states, the "gene mutation theory of cancer" does not necessarily predict that cancer should be quite common in newborns. Quite frankly, I don't know what Duesberg was smoking when he wrote that and don't see his rationale for arguing that. For one thing, it's not true that colon cancer is never seen in children. It's quite rare, to be sure, but in families with genetic mutations that predispose to colon cancer, the disease can appear in children as young as 5 or 6. I presume that Duesberg picked colon cancer because the genetic sequence of mutations that occur as the lining of the colon goes from dysplasia, to polyps, to noninvasive cancer, to invasive cancer was worked out so elegantly by Burt Vogelstein back in the early 1990's. In addition, it is true that patients with Down syndrome are more susceptible to certain cancers, specifically hematopoietic (blood) cancers, but it also turns out that they are less susceptible to solid tumors, thanks to high levels of the antiangiogenic factor endostatin, the gene for the precursor of which happens to reside on chromosome 21 and is expressed at a higher level because there are three copies rather than two. Second, Duesberg neglects to notice that oncogenes and tumor suppressors cause dysregulated growth and morphogenesis, the sine qua non of cancer. Although it is true that deleting some tumor suppressors results in viable mice who appear normal other than a predisposition to cancer, such potent growth dysregulation could cause embryonic lethality just as easily as aneuploidy could, depending on the specific gene.
Indeed, although it's not as obvious, Duesberg's making in essence the same mistake as Dr. Egnor: confusing selective mechanisms in germ line cells with those in somatic cells. A basic consideration of evolutionary theory makes it obvious that there would be major selective pressure against mutations that result in cancer in babies and children, because the cancer would kill the child before reproductive age. In contrast, cancer-predisposing genes that are either neutral before reproductive age or confer some advantage (as has been postulated, for example, for some mutations in the BRCA1 gene, which predisposes to breast cancer), may not be selected against and could remain in the germline at a high frequency in the population. Thus, from a strictly evolutionary perspective, it is not at all surprising that inherited cancers are rare in newborns and young children, even if we postulate, for the sake of this argument, that aneuploidy has nothing to do with cancer and the "genetic mutation theory" of cancer is 100% correct. Duesberg should know this, given his invocation of selective pressures leading to increased aneuploidy in cancer.
Finally, Duesberg tries to argue that mutations don't occur at a sufficient frequency under normal conditions to account for the increasing rate of cancer with age, but does not address the shortening of telomeres or that it may only take one or two mutations in key genes, such as those involved in DNA damage repair, to make the a single cell deficient in repairing its own DNA, which can then lead to an increased mutation rate, leading both to mutations and increased aneuploidy. Indeed, it has been postulated that the prerequisite for some cancers is what is termed a "mutator phenotype," in which a much higher rate of mutation is observed. In any case, aneuploidy is easier to detect than mutations, assays for which can only look at a small fraction of the genome at one time and are not sensitive enough to detect mutated cells in a background of normal cells. In contrast, small numbers of aneuploid cells can be detected in a normal background. The bottom line is that chromosomal instability is a feature of virtually all cancers but the evidence that it is driven by primarily aneuploidy is conflicting and nowhere near the slam dunk that Duesberg seems to think; in essence, it's unclear whether it is primarily aneuploidy that drives mutation or primarily mutation that leads to aneuploidy.
Here's the next bad argument:
Carcinogens take a very long time to cause cancer. Numerous chemicals and forms of radiation have been shown to be carcinogenic in animals or established as the source of occupational or accidental cancers in humans. But even the strongest carcinogens at the highest survivable doses never cause cancer right away. Instead the disease emerges only after delays lasting years or even decades. In contrast, when substances known to cause gene mutations are administered to bacteria, the cells begin displaying new phenotypes within hours; in larger organisms such as flies, the effect is seen within days. A gene mutation scenario therefore does not explain why cells exposed to carcinogenic agents become cancer cells...
Does anyone see the flaw in an argument comparing humans to bacteria or flies in this manner? Let's look at flies, because they are eukaryotes. The average lifespan of, for example, Drosophila is much shorter than a human's, on the order of 30 days or so. Carcinogens generally require cellular replication before cancer can develop. So, let's see, a latency period for cancer after exposure to carcinogens of few days in the life of a fruit fly like Drosophila is not unlike a latency period of a couple of decades in a human, if you compare it to the organism's overall life span. Bacteria reproduce amazingly rapidly; so it is not surprising that they respond to chemicals even faster. As for strong carcinogens not causing cancer right away, nothing in the genetic mutation theory of cancer demands that they must, particularly given that strong doses may result in more deleterious mutations and that the ability of a normal cell to repair its own DNA is quite prodigious. It may ultimately be shown experimentally to be true that aneuploidy is a better explanation for the long latency period of human cancers, but there is nothing in the mutation theory that demands that there must be a short latency period after a tumor cell is exposed to a carcinogenic agent, especially since it is now understood that "multiple hits" are usually required occurring in a single cell to result in cancer.
Here's the next Duesberg argument:
Carcinogens, whether or not they cause gene mutations, induce aneuploidy. Scientists have looked for the immediate genetic effects of carcinogens on cells, expecting to see mutations in many crucial genes, but instead have found that some of the most potent carcinogens known induce no mutations at all. Examples include asbestos, tar, aromatic hydrocarbons, nickel, arsenic, lead, plastic and metallic prosthetic implants, certain dyes, urethane and dioxin. Moreover, the dose of carcinogen needed to initiate the process that forms malignant tumors years later was found to be less than one-thousandth the dose required to mutate any specific gene. In all cases, however, the chromosomes of cells treated with cancer-causing doses of carcinogens were unstable--that is, displaying higher than usual rates of breakage and disruption.
Sure, carcinogens induce aneuploidy, but just because some carcinogens do not directly damage DNA does not necessarily mean that the induction of aneuploidy must be the mechanism by which they cause cancer. It might be, but it doesn't necessarily have to be. For example, part of the mechanism by which asbestos is thought to cause cancer is by causing chronic inflammation, which chronically stimulates nearby cells to divide, while at the same time exposing them to a milieu containing numerous inflammatory cytokines secreted by white blood cells that invade the area. In the case of dioxin, for example, there appears to be a receptor to which dioxin binds that stimulates cell survival pathways. Either of these mechanisms, and others, could be at play in the carcinogenesis due compounds that do not cause extensive DNA damage and could account for the long latency period between exposure to carcinogen and cancer.
The rest of Duesberg's arguments range from the "so what?" to the more intriguing. For example, he points out that different patterns of aneuploidy are seen in different tumors. Given the known genetic heterogeneity of cancers, this is not at all surprising and doesn't necessarily mean that aneuploidy is the primary cause of cancer. For example, the Philadelphia chromosome, the result of a reciprocal translocation, an exchange of genetic material, between chromosomes 9 and 22. It is very common, virtually pathognomonic for chronic myelogenous leukemia. This is associated with the excessive production of an oncogene (Bcr-Abl), which activates cell cycle genes and induces genomic instability. Certainly, there must be some sort of aspect to the structure of these two chromosomes that make this particular translocation as common as it is, but the end result that drives CML is a mutation that results in the excessive production of an oncogene. Indeed, molecularly targeting this oncogene has resulted in a very effective drug against CML called Gleevec. In fact, targeting single gene abnormalities, although it hasn't resulted in a cure for cancer, has resulted in several very effective treatments, such as Herceptin, which is directed against the Her-2/neu oncogene. Finally, lots of single gene mutations in transcription factors, DNA repair enzymes, or cell signaling molecules can alter the expression of dozens or hundreds of downstream (or even thousands) of genes in predictable ways. Large scale chromosomal breaks are not necessary to account for such globally deranged gene expression.
Duesberg next argues that "gratuitous traits do not contribute to cancer survival," referring specifically to genes for metastasis and drug resistance, oddly enough. The problem with this argument is that a subset of the genes for carcinogenesis are not infrequently also genes involved in metastasis. Similarly, genes involved in drug resistance often have other functions than just drug resistance. For example, the mdr1 multidrug resistance gene product is an ATP-dependent channel that extrudes a variety of substances, not just chemotherapeutic agents, from a cell, an excellent example of how evolution can coopt the function of an existing protein to do another function. Finally, Duesberg appeals to the ability of cancer cells to change their phenotype rapidly, supposedly much faster than genes can mutate. This may indeed be true, and it may indeed be possible that aneuploidy contributes to this, given its ability to cause wholesale rearrangements of chromosomes and hundreds (or even thousands) of genes in one fell swoop. However, all it takes is selection by drug for tumor cells that express ever-increasing amounts of mdr1; chromosomal rearrangements, although they certainly may contribute to drug resistance, do not appear to be strictly necessary for it. Moreover, once again, Duesberg cannot resist overselling his case:
Once this vicious cycle is under way, the fact that every cell would be randomly generating its own new phenotypes could explain an observation made decades ago by Leslie Foulds of the Royal Cancer Hospital in London that "no two tumors are exactly alike ... even when they originate from the same tissue ... and have been induced experimentally in the same way." Such individuality is yet another hallmark of cancer that cannot be explained by the activity or inactivity of specific genes, which would be expected to have consistent effects each time and in each cell.
The problem with this argument is that it's only partially correct. Using the "gene chip," which now allows scientists to assay the levels of every gene in the human genome at the same time, we now know that tumors often have a surprisingly similar pattern of expression of thousands of genes. Indeed, one of the most startling findings early in the use of such gene chips was that nearly all breast cancer could be divided on the basis of gene chip experiments into a small number of distinct subtypes, the main ones of which include the "basal" (more aggressive) or "luminal" (less aggressive) phenotype. Moreover, the expression of the Her-2/neu oncogene leads to a distinct, identifiable, reproducible gene expression pattern, in direct contradiction to Duesberg's claims above. Indeed, tests based on such gene chips are already making their way into the clinic to estimate a patient's risk of recurrence and guide chemotherapy decisions.
Lest one think that I'm hostile to Duesberg's hypothesis, let me disabuse you of the notion right now. Although I think Duesberg's an utter crank and pseudoscientist when it comes to his HIV/AIDS denialism, I find some of his work in cancer intriguing, and I disagree with Mark and Larry that it was such a horrible thing to feature him in an article in Scientific American, especially given the disclaimer. It is clear to me that epigenetics (cellular factors other than genes that regulate gene activity) and chromosome structure are very important in carcinogenesis, more so than had been appreciated before. However, contrary to how Duesberg's sycophants like to portray the chromosomal hypothesis of cancer as an epic battle that's all about Duesberg, who is portrayed as the lone voice arguing for the hypothesis that aneuploidy is the cause of cancer, in reality it's nothing more than yet another scientific controversy that is, fortunately, no more nasty than a lot of other controversies in science, such as, for example, the hypothesis that changes in cell metabolism are the cause of, not a consequence of, carcinogenesis. It's also nowhere near as clear as Duesberg claims whether aneuploidy is a cause or a consequence of carcinogenesis. For one thing, there are at least two examples that I'm aware of (which means there are probably more than that) of groups generating tumor cells that do not have significant or widespread aneuploidy, demonstrating that aneuploidy may not be a prerequisite for carcinogenesis, in direct conflict with Duesberg's hypothesis. In addition, there's a very interesting article from Don Cleveland's lab in the January Cancer Cell that suggests that aneuploidy can promote carcinogenesis under some circumstances (an observation that seems supportive of Duesberg's hypothesis) and act as a tumor suppressor under others (an observation that is arguably not).
What really irks me about Duesberg with respect to his ideas about cancer is that he may be on to something, but he can't seem to stop himself from the same black-and-white, either-or thinking that apparently led him down the road of HIV crankery, nor can he seem to resist massively overselling his hypothesis as the be-all and end-all hypothesis to explain cancer initiation and progression. As I said at the beginning of my post, whenever someone postulates theirs as The One True Cause of Cancer, my skeptical antennae start twitching, and Duesberg's aneuploidy hypothesis is no exception. Cancer is a complex and resourceful foe, not to mention that it's hundreds of different diseases, not a single disease. Duesberg neglects a variety of other new hypotheses for causes of carcinogenesis that hold equal or greater promise than the chromosomal chaos hypothesis. Among these are cancer stem cells, tumor angiogenesis, and the aforementioned metabolic hypothesis of cancer (a.k.a. the Warburg effect). He even neglects what I consider to be a far more fascinating and sophisticated version of the chromosomal hypothesis, specifically Tom Misteli's concept that derangements in the higher order three dimensional structure of chromosome territories can lead to cancer by alterations in gene expression.
Duesberg's supporters may look at the relative neglect of chromosomal structure as a controller of gene expression and a potential cause of cancer and wonder why it was neglected for so long. The analogy I like to make is to politics. It is said that politics is the art of the possible. To me, science is the study of what it is possible to study. Two to three decades ago, we figured out how to study individual genes; so that's what we studied, even though we soon realized that such reductionist techniques did not give the complete picture of cancer. Less than 10 years ago, gene chips, coupled with improvements in statistical analysis and increases in computing power that made it possible to analyze the data produced from such huge experiments, allowed us to look at the expression of the entire genome at once, leading to a richer understanding of the changes in gene expression that occur during cancer. The more sophisticated techniques and understanding did not invalidate what we had learned before; it complemented and extended it. Similarly, we now have the tools to probe chromatin structure at a level of detail never before possible; consequently we are now looking at chromatin structure in cancer. Our increasing ability to probe the detailed structure of chromosomes will likely now complement and extend what we have learned about cancer through the study of mutations in individual genes. Walter Giaretti asks:
...I would like to pose the question if the "aneuploidy theory" of cancer in relationship with the "mutation theory" still remains as controversial as in the near past. Don't we have now enough experimental evidence that cancer originates and progresses with the contribution of both gene mutations and aneuploidy?
Duesberg's failure is that he doesn't seem willing to accept that the answer to this question is almost certainly "yes." As Giaretti puts it:
It is likely that new studies directly comparing DNA copy number and gene expression will be performed in the near future on the role of aneuploidy in cancer, on what genetic events may induce chromosomal instability and on the validation of novel criteria for early diagnosis. It is predictable that these studies will vanish the conflicting views that either aneuploidy or gene mutations are a unique cause of the origin and progression of cancer negating the role of the alternative mechanism. Today, these conflicting interpretations are increasingly being abandoned to let a more complex mixed paradigm take over from previous concepts. In brief, ideas stemming from the old Boveri theory and from the modern theories may soon be seen as cooperative and equally important to cancer.
There are indeed deficiencies in our current understanding of cancer initiation and progression, but there's no reason that gene mutations and aneuploidy couldn't both contribute to these processes. Indeed, I'd be surprised if it were otherwise. Duesberg seems too dogmatic and wedded to his hypothesis to see the big picture.
Just how central is Duesberg to the latest research in aneuploidy and cancer. I read Bialy whining somewhere that Duesberg was not cited in a recent review of aneuploidy and cancer. I get the impression that Duesberg (or at least Bialy) is overselling his role. Certainly Duesberg's sycophants seem to believe that Duesberg is the centre of the world.
The one thing that Duesberg is doing that others aren't is taking an extreme position.
Gene mutations, aneuploidy, epigenetic processes, tumor environment and anything else that alters gene expression. Duesberg is far too dogmatic.
The cynic in me thinks that flogging Duesberg's role in popularizing interest in the role of aneuploidy in cancer may just be a way of drumming up business for this company that he, Bialy and Rasnick are associated with that is marketing or attempting to market this Anucyte cancer detection system? http://www.primenewswire.com/newsroom/news.html?d=112649
Isn't Down syndrome due to a non-disjunction event in the second meiotic division, not mitosis? In that sense, it's different than aneuploid cancer lineages which arise due to mitotic non-disjunction.
I may have missed this, but is the main chromosomal abnormality in Duesberg's article aneuploidy? What about other aberrations (fusions, translocations, inversions, duplications, deletions, etc)? Aren't there a lot of cancers associated with chromosomal defects other than aneuploidy?
Retinoblastoma.
Not to be too cryptic, but as far as kids not getting cancer? Nope.
As far as chromosomal defects yes. In particular, it's not the aneuploidy of a huge number of translocations seen in leukemias and lymphomas but the creation of fusion genes and inappropriately expressed genes from the transformation (a promiscuous promoter driving an oncogene for instance) that results in specific types of cancer.
Orac is right. Duesberg is being to simplistic and rigid.
Acute promyelocytic leukemia.
The majority of cases of acute myelogenous leukemia.
Fixed.
In these epic posts, the occasional screwup does occur. Mea culpa. It's kind of like the scene in Animal House:
I was on a roll.
Orac,
Thanks for a typically smart and insightful criticism of Duesberg's article, not to mention an admirably reasonable reaction to the fact of Duesberg's authorship.
At the risk of seeming unappreciative: Scientific American's lawyers do usually frown on the use of its graphics without permission--strictly speaking, it isn't a violation of our rights so much as it is of the artist's. I don't think she'd mind in this case, but can I offer a shout of appreciation to the terrific Jen Christiansen, who drew that illustration?
Finally got to read the whole thing, it was a bit epic.
I agree with you, this almost seems to be a presage to even more crankery from Duesberg. I can just see that as time goes on and this hypothesis is balanced out by other findings that show every cancer can't simply be housed under the umbrella of aneuploidy if he'll once again just dig in and refuse to accept evolving facts. When Duesberg suggested that HIV causing AIDS was based on mostly correlative evidence in 1987 (and Science published it), he kind of had a point. It wasn't a particularly good, or useful point, but within the year there was excellent evidence from studies of AZT, as well as responses to Duesberg's criticisms that really should have had those ideas tossed, and certainly by about 1993 or so the science was settled. These days given what we know about the HIV proteins, mechanisms of latency and entry in CD4 cells, it's frankly just embarrassing he didn't give up decades ago, I wonder if he'll do the same here if things don't turn out his way.
I will continue to disagree, however, that this wasn't a mistake for Sci Am. I believe in the scientific death sentence - that is, if you are proven untrustworthy, you shouldn't be allowed to contribute to the literature anymore. So what if he's changed fields, he demonstrated himself to be a dishonest crank so badly and for so long why should we risk the literature with contributions from this guy? I don't find his so-called dissident status cute or funny, it results in bad science, bad policy and ultimately death as these bizarre ideas take hold in Africa (especially S. Africa) and even in the United States as with the HARM people. I'm all for the black list for cranks too because I believe cranks are fundamentally deceptive and untrustworthy contributors to scientific debate, and all of his subsequent (thankfully rejected) internet publications on HIV/AIDS are really just a pack of lies.
I simply wouldn't trust this guy again, ever.
John Rennie,
I salute you, Sir, for publishing the article by Dr. Peter Duesberg on aneuploidy and cancer. His hypothesis is clearly testable: produce a diploid tumor, somewhere, somehow.
I disagree, though, that Orac's criticism is "smart" or "insightful." Rather, it is a mess. It could never be published anywhere -- he intersperses irrelevant issues (Egnor and creationism), spends half the time talking about Duesberg and his supporters, carping on strawman HIV issues, and in typical verbose fashion misses the import of the issue.
Cancer kills over 500,000 Americans each year. Most families have lost a loved one to cancer. Aside from the success in lung cancer (smoking reduction) and some leukemias (a few new drugs), cancer prevalence rates haven't budged since the 1950s.
So, what clinical benefits or societal benefits has the "oncogene" theory of cancer produced in nearly 40 years? I submit --zero.
That's why the Duesberg article is important -- old wine in a new bottle as Nature Biotechnology once wrote.
Let's not lose sight of the objective -- to cure cancer. To cure something, one has to properly determine its cause. Since aneuploidy, apparently, is found in every solid tumor, it certainly must be explained -- not explained away as Orac in typical smarmy fashion seeks to do.
Sanderson, you nut-job, why not read what Orac wrote before you piss on it? Then you would know that Orac doesn't explain away aneuploidy at all. He actually supports Duesberg on this one.
You might also learn something about cancer. Like about Herceptin and Gleevec, drugs that wouldn't work at all if there weren't some truth to the oncogene hypothesis.
Why don't you go and hang out with your three denialist friends. Maybe you can teach each other how to read.
In my opinion, what has driven interest in the role of genomic instability and aneuploidy in cancer isn't Duesberg but the technological advances of the early 90s (like fluorescent in situ hybridization) that have enabled investigators to examine ploidy in tumors in far more detail than was possible prior to the 90s. Also my opinion - aneuploidy and mutations both contribute to cancer but aneuploidy is more obvious in tumors than mutations are because it's easier to detect.
I'm not sure what the fine print of the "fair use" law says, but I really feel that Orac is ripping SciAm off a bit here. From my vantage point, I'd fully support a recommendation from SciAm that Orac remove the pictures in this blog. They go beyond fair use, unless Orac's post was along the lines of "SciAm used artwork which gives a misleading impression..." (that is, the pictures were a cornerstone of the point Orac was making).
I think that a more appropriate treatement from Orac would be a summary of the article with perhaps a selected quote or two, and a comment along the lines of "the ideas are very elegantly presented in artwork accompanying the SciAm article" (with a link to the paid-for article).
Sadly typical of Duesberg. Can't accept that the concept isn't exactly novel, can't accept that other theories have validity, can't accept supporting role instead of leading man.
Dear Dr.
For a different view of cancer, you might want to see the article shown on the cover of The American Journal of Pathology in this month's MAY issue. (http://ajp.amjpathol.org/) (Tone Sandal, Klara Valyi-Nagy, Virginia A. Spencer, Robert Folberg, Mina J. Bissell, and Andrew J. Maniotis.
Epigenetic Reversion of Breast Carcinoma Phenotype Is Accompanied by Changes in DNA Sequestration as Measured by AluI Restriction Enzyme. Am J Pathol 2007 170: 1739-1749.) I'll send you the entire PDF if you send me an address where I can email it.
I read your assessment of Peter's SCi Am. article on aneuploidy, and agree with you about the excitement regarding the renaissance of global changes in higher order structure, and cancer, that similar to aneuploidy, involve entire genomic structural changes. You might want to look at what others as well as I have written regarding this issue of "mechanogenomics," as I called it.
Stein GS.Am J Pathol. Mechanogenomic control of DNA exposure and sequestration. 2005 Apr;166(4):959-62.
Maniotis AJ, Valyi-Nagy K, Karavitis J, Moses J, Boddipali V, Wang Y, Nuñez R, Setty S, Arbieva Z, Bissell MJ, and Folberg R: Chromatin organization measured by Alu I restriction enzyme changes with malignancy and is regulated by the extracellular matrix and the cytoskeleton. Am J Pathol 166: No. 4 April 2005.
Folberg R, Arbieva Z, Moses J, Hayee A, Sandal T, Kadkol S, Lin AY, Valyi-Nagy K, Setty S, Leach L, Chevez-Barrios P, Larsen P, Majumdar D, Pe'er J, Maniotis AJ.
Tumor cell plasticity in uveal melanoma: microenvironment directed dampening of the invasive and metastatic genotype and phenotype accompanies the generation of vasculogenic mimicry patterns. Am J Pathol. Oct;169(4):1376-89, 2006.
Valyi-Nagy K., Folberg R., Valyi-Nagy T. Andrew Maniotis. Role of tumor invasiveness, the extracellular matrix, and chromatin sequestration in the susceptibility of uveal melanoma to herpes simplex virus type 1. Experimental Eye Research 84 (2007) 991-1000.
Mechnogenomic control of DNA exposure and sequestration.
Drawings from 100 years ago of Galeoti's, Hertwig's, and Hanseman's comparisons of cancer cell and normal cell chromosomes illustrated gross differences in chromatin structure and chromosome numbers in human tissue harboring a tumor (1). But how higher order chromatin structure is controlled and maintained by the cancer cell and its microenvironment has remained beyond our reach. The work presented by Maniotis et al. in this issue presents new data that shows there are striking differences in restriction enzyme sensitivity between tumor cell and normal cell genomes. The work shows in addition, that these differences depend upon chromatin-associated proteins rich in disulfide bonds, specific molecules present in the extracellular matrix (ECM) environment, and the cytoskeleton, whose organization permits or impedes sequestration and exposure of specific sites along DNA. The work advances the idea that the mechanical contiguity of these components may work in concert to form a cytoarchitectural resistance mechanism that may constitute a basis for a paradigm shift in both the contexts of normal cellular physiology, and in cancer.
It had been established by a previous generation of tumor biologists using DNase digestions and nick-end labeling techniques, that in order for a gene to become expressed, it had to be "exposed." Moreover, Puck et al., (2) had shown that transformed tumor cells that were "reverse transformed" to a normal phenotype with various chemical compounds exhibited a shift in their nuclei's sensitivity to DNAase I, and also exhibited changes in cell shape and cytoskeletal organization.
Maniotis et al. in this issue show data that compared the extent of exposure or sequestration of a well-characterized collection of highly invasive, poorly invasive, and normal human cell genomes to specific restriction enzymes. Restriction enzyme digests on intact genomes untreated with other chemical agents showed that Alu I and Msp I both could distinguish normal cells from tumor cells of different degrees of invasiveness. Highly invasive cell nuclei always resisted digestion with these enzymes compared to poorly invasive or normal cells. Also, through comparisons of mitotic chromosomes microsurgically extracted as complete genomes (3,4), with interphase nuclei from the same cell types, differences in chromatin digestibility were shown to be independent of the cell cycle, and localized to chromosomes, without the possibility of enzyme access contributing to the differential digestive effects observed in different cell types. The different relative sensitivities of chromatin or chromosomes to restriction enzymes was also demonstrated to be independent of ploidy, but sensitive to oncogene insertions on the same genetic background. Moreover, the differences in chromatin and chromosome stability in comparisons of highly invasive, poorly invasive, and normal cells was also found to involve proteins rich in disulfide bonds. This phenomenon was demonstrated by adding DTT or Ã-mercaptoethanol to the digestion reactions and rendering the highly invasive cell genomes sensitive to Alu I.
In the second part of the work, a systematic search was initiated to test if growth factors, or soluble and polymerized ECM molecules also induce cells to alter their sequestration or exposure of DNA, and to test if there was any specificity to the influence of different ECM molecule types on Alu site exposure or sequestration. These experiments showed that soluble laminin and RGD-C were both capable of rapidly and profoundly causing all cell types that were tested to sequester their DNA from the digestive effects of Alu I. However, sequestration induced by these ECM molecules was always more intense among increasing grades of cellular invasiveness. Serum, fibronectin, bFGF, EGF, and type I collagen exerted no effect on the seqestration of Alu I sensitive sites. The same results were obtained with polymerized extracellular matrix conditions. In addition, when polymerized ECM preparations were prepared such that ECM molecules were deposited in specific regions of a surface, the contrast in chromatin sensitivity could be easily observed between cells touching the extracellular matrix compared to the same cells that did not touch ECM deposits.
These studies suggest several novel biological concepts: through comparisons of the sensitivities of chromosomes derived from a chromatin extraction method that preserves the native structure of chromatin with digestion of specific Alu I and Msp I sites with digestions of interphase cells of the same type that are exposed or not exposed to defined matrices, a more accurate picture is provided as to how global chromatin structure exists and is controlled under physiological ion conditions, and how the organization, mechanics, and function of different types of genomes are linked to DNA sequestration, and exposure, and how these processes might occur at the level of tissue.
These findings have special relevance in tumor biology. It is now known that laminin is a principal component of extravascular matrix-rich vasculogenic mimicry patterns, or fluid-conducting meshworks that conduct plasma within tumors that probably arises from leaky vessels disturbed by invasive tumor cells (5-11), and whose presence designates how patients harboring melanomas and other tumors will progress (see Ref. 12). The fact that laminin and RGD-C sequester the Alu sites in both normal and tumor cells in only 30 minutes is remarkable because it suggests that specific ECM moieties present in the environments of malignant tumors can rapidly and profoundly induce a complete reorganization of the higher order structure of the genome to sequester Alu sites. However, these observations also suggest that perfusion itself, by whatever route (eg. tumor angiogenesis, vasculogenic mimicry, vessel cooption, etc.) may be a small part of the story regarding how factors get into cells of highly invasive tumors. These data would suggest that the highly invasive cells themselves, owing to their abnormal organizational characteristics, may be the principal directors for both nutrient access, and many of the nucleic acid-disrupting drugs used in cancer chemotherapy.
Furthermore, it is clear that conceptual frameworks advanced to explain how prokaryotic genomes are controlled cannot explain how the regulatory machinery controlling higher order chromatin structure coordinates large regions of the eukaryotic genome along with its batteries of hundreds, or perhaps thousands of genes and gene networks. For instance, it has been established that even among single cell eukaryotes such as yeast, the majority (>95%) of single-gene mutations in yeast affect not only the expression of the mutant gene, but also the expression of many other genes (13).
In multicellular organisms, in addition, the cytoskeleton and extracellular matrix (ECM) are known to play a fundamental role in determining cellular behaviors. To test if different cytoskeletal fiber systems influenced the sequestration or exposed the Alu I sites, a variety of cytoskeleton-disrupting drugs were then employed to determine if the higher order structure of chromatin was controlled by actin, microtubules, or intermediate filaments. It had been previously established that a tug to an integrin receptor could alter the molecular alignment of intranuclear molecules in 1 second, and each cytoskeletal system had been shown to exert different stabilizing effects on both nuclear structure, and force transduction (14). The results of these experiments showed that indeed, as with the force-transduction experiments on integrin receptors, each cytoskeletal fiber system contributed profoundly to the sequestration or exposure effect, such that actin disruption decreased sequestration, while microtubule or intermediate filament disruption increased sequestration.
These findings raise and experimentally address new questions regarding the fundamental way genetic information is controlled by proteins containing disulfide-rich bonds, by the extracellular matrix microenvironment, and by the cytoskeleton. The work also has contributed several new approaches that can be used as diagnostic tools that use the differences in the sensitivities of DNA to distinguish differing degrees of malignancy that do not depend on molecular markers which have been shown to be highly variable in the context of vasculogenic mimicry, and which manifests as molecular mimicry in tissue sections of the most invasive types of tumors (5,15,16). These factors working separately, or in concert, may have relevance to the "resistance" of malignant cells, and have implications regarding how drug resistance of the most malignant types of tumor cells may be induced by the presence of specific types of extracellular matrix moieties. It appears the architecture of cytoplasm and nuclei themselves, induced by the extracellular matrix, dictates how DNA is sequestered or exposed. Models of higher order chromatin structure and gene control in higher eukaryotic cells must be considered contextual and hierarchical with respect to the ECM environment and the cytoskeleton (17). In this context, and as suggested by the authors, tensional integrity (tensegrity) may be a useful conceptual framework to account for how molecules can function collectively as components of integrated, hierarchical systems, in the physical context of living cells and tissues (18).
References
1. Wilson EB. The Cell in Development and Inheritance. Pathological mitosis in cancer cells, pps. 68,69. Reprinted from the New York Edition of 1896. The sources of Science # 30, Johnson Reprint Corporation, New York and London, 1966.
2. Puck TT, Krystosek A, Chan DC. Genome regulation in mammalian cells. Somat Cell Mol Genet 1990 16: 257-265.
3. Maniotis, A., Bojanowski, K., Ingber, D. Mechanical continuity and reversible chromosome disassembly within intact genomes removed from living cells. J. Cellular Biochem. Vol 65: 114-130, 1997.
4. Bojanowski, K., Maniotis, A., Ingber, D. DNA toposiomerase ll can control chromatin topology and drive chromosome condensation without enzymatically modifying DNA. J. Cellular Biochem. Vol. 69:127-142, 1998.
5. Maniotis A., Folberg R., Hess A., Seftor E., Gardner L., Pe'er J., Trent J., Meltzer P., Hendrix M. Vascular channel formation by human uveal melanoma cells in vivo and in vitro: Vasculogenic mimicry. Amer. J. Path. Vol. I55, No 3, pps. 739-752, September,1999.
6. Andrew Maniotis, Xue Chen, Christopher Garcia, Phillip J. DeChristopher, Ding Wu, Jacob Pe'er, Robert Folberg. Control of Melanoma Morphogenesis Endothelial Survival, and Perfusion By Extracellular Matrix. Lab Investigation. Vol. 82 No. 8 p.1083-1092, 2002.
7. Shirakawa K, Tsuda H, Heike Y, Kato K, Asada R, Inomata M, Sasaki H, Kasumi F, Yoshimoto M, Iwanaga T, Konishi F, Terada M, and Wakasugi H. Absence of Endothelial Cells, Central Necrosis, and Fibrosis Are Associated with Aggressive Inflammatory Breast Cancer. Cancer Research 61, 445-451, January 15, 2001.
8. Shirakawa K, Kobayashi H, Heike Y, Kawamoto S, Brechbiel M, Kasumi F, Iwanaga T, Konishi F, Terada M, Wakasugi H. Hemodynamics in Vasculogenic Mimicry and Angiogenesis of Inflammatory Breast Cancer Xenograft. Cancer Research 62, 560-
566, January 15, 2002.
9. Kobayashi H, Shirakawa K, Kawamoto S, Saga T, Sato N, Hiraga A, Watanabe I, Heike Y, Togashi K, Konishi J, Brechbiel MW, Wakasugi H. Rapid accumulation and internalization of radiolabeled herceptin in an inflammatory breast cancer xenograft with vasculogenic mimicry predicted by the contrast-enhanced dynamic MRI with the macromolecular contrast agent G6-(1B4M-Gd)(256). Cancer Res. 2002 Feb 1;62(3):860-6.
10. Shirakawa K, Kobayashi H, Sobajima J, Hashimoto D, Shimizu A, Wakasugi H. Inflammatory breast cancer: vasculogenic mimicry and its hemodynamics of an inflammatory breast cancer xenograft model. Breast Cancer Res. 2003;5(3):136-9. Mar 06, 2003.
11. Clarijs R. Otte-Holler I, Ruiter, de Waal MW. Presence of a fluid-conduting meshwork in xenografted cutaneous and primary human uveal melanoma. Investigative Ophthalmology and Visual Science: Vol. 43 No 4 2002.
12. Folberg R, Maniotis AJ. Vasculogenic mimicry. Acta Pathologica, Microbiologica, et Immunologica, Scandinavica. Jul;112 (7-8):508-525,. 2004.
13. Featherstone DE, Broadie K. Wrestling with pleiotropy: genomic and topological analysis of the yeast gene expression network. Bioessays. Mar;24(3):267-74, 2002.
14. Maniotis, A., Chen, C., Ingber, D. Demonstration of mechanical interconnections between integrins, cytoskeletal filaments, and nuclear scaffolds that stabilize nuclear structure. Proc. Nat. Acad. Sci . Vol. 94 pp.849-854, 1997.
15. Chen X, Maniotis AJ, Majumdar D, Pe'er J, Folberg R: Uveal melanoma cell staining for CD34 and the assessment of tumor vascularity. Invest Ophthalmol Vis Sci. Aug; 43(8): 2533-9. 2002.
16. Seftor EA, Meltzer PS, Kirschmann DA, Pe'er J, Maniotis AJ, Trent JM, Folberg R, Hendrix MJ. Molecular determinants of human uveal melanoma invasion and m
metastasis. Clin Exp Metastasis. 2002;19(3):233-46.
17. Roskelley CD, Srebrow A, and Bissell MJ: A hierarchy of ECM-mediated signaling regulates tissue-specific gene expression. Curr Opin Cell Biol 7: 736-747, 1995.
18. Ingber DE. The Architecture of Life. Scientific American, January, pp 48-57, 1998: see also Ingber DE. Tensegrity II. How structural networks influence cellular information processing networks. Journal of Cell Science 116, 1397-1408, 2003.
Trying to blow your own trumpet again, Andrew? Perhaps more people might read what you write if you presented it in smaller digestible chunks, and more might listen to what you have to say if you dropped the dogmatic and anti-scientific approach that you share with Duesberg in refusing to be open to alternative hypotheses and willing to accept when you have been shown to be wrong (as you both have in relation to your AIDS denial).
Wow, Dr. Maniotis, nice work! I just have to wonder how you justify your work with "Alu I". Because I personally don't believe it exists.
Do you have electron micrographs of Alu I in the act, cutting DNA? Have you confirmed this with crystal structures? Are your Alu I preps 100% pure? Are you sure the DNA isn't just being cut by endogenous restriction enzymes activated by oxidative stress when you add your inevitably impure "Alu I"? Did you check the redox environment of the cells? Isn't it possible that the DNA in cancer cells is more sensitive to "Alu I" digestion because of their different redox state?
Another thing I notice is you use the T4-2 breast cancer cell line. You know better than anyone, "to culture is to alter." So what's the relevance? Why didn't you do this work in situ in patients? Cancer has been around forever, and there's still no work on Alu I sensitivity directly in patients? Unbelievable.
Obviously, AluI doesn't exist. And now I'm not even sure about cancer.
Methinks Dr. Maniotis would like me to blog his latest article. A little bit excessively self-promoting, but what the heck? I might even be willing to oblige him as long as it's not a Wiley & Sons publisher, where I might have to worry about being sued if I were to do a selective quotation or two or appropriate a smaller version of one of his figures for "fair use." I'm always happy to talk science, as opposed to pseudoscience like HIV/AIDS denialism, and I'm equally happy that Dr. Maniotis refrained from mentioning his views on HIV/AIDS, given that he is a signatory to a letter by the Perth Group stating that HIV doesn't cause AIDS.
As long as we stick to cancer, I don't see why we couldn't have a fruitful discussion, and I don't see why I couldn't have a fruitful discussion even with Duesberg himself. For one thing, I'd be interested in knowing Dr. Maniotis' thoughts on Dr. Duesberg's arguments, as presented in the SciAm article and a couple of recent scientific reviews, particularly bad arguments such as "babies don't get cancer."
I would like to see a fruitful discussion on cancer between Orac and Dr. Maniotis. However, could you shorten it a bit? Not every scientific argument has to be "War and Peace"
Adele, As you are no doubt aware there are still ethical questions about the circumstances under which Watson and Crick obtained Rosalind Franklin's crystallographic data of DNA structure. I think to be on the safe side you should add DNA to your list of things whose existence must be questioned.
Orac:
You state that would like to address "particularly bad arguments such as "babies don't get cancer." But, Dr. Duesberg didn't write that and you know it.
He wrote:
Lamentably common, cancer afflicts about one in three people at some point in their lives, but mostly after the age of 50, which is when chances for malignancy soar. Thus, cancer is, by and large, a disease of old age.
How can anyone dispute this? He then wrote:
But colon cancer is never seen in children., which you deliberately misinterpreted this narrow quote to include cancer in general.
So, back to the properly framed issue:
If you can produce babies with colon cancer, then your point is well-taken and Dr. Duesberg is in error. If not, then your point is not well-taken, and you are in error.
Here's a paper from the NCI on colon cancer.
It's pretty good, but doesn't mention any babies getting colon cancer, so it doesn't quite answer the question.
Here's a SEER Fact Sheet, also from the NCI. It states:
From 2000-2004, the median age at diagnosis for cancer of the colon and rectum was 71 years of age. Approximately 0.0% were diagnosed under age 20.
So, according to these authorities, Duesberg is right: babies don't get colon cancer. At least from 2000 - 2004. Do you have any comment on these facts?
Louie,
Duesberg is wrong on colon cancer in children. They do get colon cancer, if rarely.
Colon cancer anyway was just Duesberg's example for cancer in general. His mouse comment makes that obvious. So Duesberg makes it clear he believes cancer doesn't happen in children unless congenital aneuploidy is present.
I don't know for sure, but I think it's just the opposite. Most pediatric cancers are familial with inherited mutations in DNA repair genes. And those cancers may not be aneuploid (look at the review Rajagopalan and Lengauer Nature 2004).
Duesberg's ideas about cancer and his HIV denialism are materially connected: Duesberg's conference on cancer and aneuploidy in San Francisco in 2004 was underwritten by Robert Leppo, a right wing Republican venture capitalist and denialist who was also the executive producer of the film "The Other Side of AIDS," made by Robin Scovill and featuring his wife, HIV+ Christine Maggiore: Scovill and Maggiore allowed their 3 year old daughter to die, untested and untreated, of AIDS in 2005. Leppo also bought the building used by the AIDS denialist fringe group ACT UP/SF, and was a member of South Africa's President Thabo Mbeki's notorious AIDS panel.
Deusberg and his long-time sidekick, David Rasnick PhD, are in business together with an aneuploidy test they claim will detect all kinds of cancers. Rasnick, of course, was also influential in shaping South Africa's President Thabo Mbeki's views about AIDS and thus Mbeki's deadly policy of refusing to provide South Africans with AIDS access to antiretroviral drugs, and until recently worked for Mathias Rath, the vitamin quack/magnate/denialist (see the recent article by Michael Specter in The New Yorker for more on Rath and Rasnick). Their company was called Boveran, and the test was called "iCyte" until they ran into some trademark issues.
In October 2006, Rasnick and Duesberg sold Boveran to Modern Technology Corp, a shady little biotech company (and alpaca farm) in Mississippi trading as a penny stock under the symbol MODC--and steadily losing shocking amounts of its investors' money. The CEO Anthony Welch seems to be hiding offshore somewhere--possibly Jamaica, possibly the Bahamas. The corporation is registered in Nevada, which boasts that it offers the strongest protections from lawsuits of any state. (For some interesting background to MODC by a technology writer named Julie Jacobson see www.cepro.com/news/editorial/7779.html.) Boveran was renamed Insight Medical Group, and is a wholly owned subsidiary of MODC; Duesberg and Rasnick are still the primaries. Insight Medical Group is establishing its cancer diagnostics lab in Freeport, Grand Bahama Island (!). This way they can avoid the bureaucracy of clinical trials and FDA approval.
Just last month (on March 29, 2007), Modern Technology Corp announced that Mexico-based ex-scientist Harvey Bialy, an exceptionally bizarre and floridly homophobic member of the denialist cadre, had joined Insight Medical Group's medical advisory board for the cancer test, now renamed "Anucyte." Bialy wrote a hagiographic biography of Duesberg and was recently "retired" from his last academic affiliation at UNAM in Mexico. (For more on Bialy, go to www.AIDStruth.org and look for "homophobic" on the menu on the upper left.)
Another recent addition to Modern Technology Corp's cadre of experts is an eye doctor named Marc Rose, who was expanded his interests from sight preservation to male menopause and "life extension": he is active with the Cancer Control Society, which among other things organizes bus tours of alternative cancer clinics in Tijuana, Mexico. He will work on the further development and marketing of the Anucyte test.
The CEO of Modern Technology Corp is Anthony K Welch, who studied Electrical Engineering for 2 years (1986-88, no degree awarded as far as I can tell) at University of Mississippi, and now claims to be a law student at Concord School of Law. (Concord School of Law is an on-line school that told me that they do not accept students without degrees; it's not clear how Welch finessed that in his application to Concord's JD program.) Despite extraordinary financial losses and many complaints to the SEC from angry investors (search "MODC" on such bulletin boards as Ragingbull.com and allstocks.com for investor opinions of Welch as a scammer and a fraud), Welch has been paying himself a very hefty salary--almost $300,000 in for the last fiscal year. His CFO, Robert Church, resigned from the company last June, and Welch took over that function.
Insight Medical Group promises "to provide ongoing financial support to Peter Duesberg's lab ... [which] ...agrees to work closely with Insight Medical Group to improve products and technology" (www.primenewswire.com/newsroom/news.html?d=111522). That is to say, Duesberg is a principle of a subsidiary that has as its sole asset an offshore lab (which may or may not exist) for a cancer diagnosis technology that has not been clinically tested or approved by the FDA or any other objective institution. The CEO is regarded by investors as unreliable, the company that is at best poorly managed. Perhaps Duesberg, Bialy, Rasnick and Welch thought that Duesberg's article in Scientific American would at least temporarily inflate the value of the stock of the company that would then provide funds to Duesberg's Berkeley lab. But the SciAm article has not helped Modern Technology Corp's stock price at all--it's down to $.012 a share from almost 6 cents a share a year ago.
Indeed. Ky is being disingenuous by citing a "0.0%" rate of colon cancer under age 20. As the reference states, it's a rounded figure. If, for example, the true rates were 0.049%, that would round to 0.0%.
Adele is correct that Duesberg is obviously choosing colon cancer primarily as an example and that he is arguing that the scarcity of cancer as "evidence" that mutation theory is incorrect and his aneuploidy hypothesis is correct. My main point (and the reason why Duesberg's argument is so bad) is that the observation that cancer is uncommon in newborns is a non sequitur. It does not follow from this observation that mutations don't cause cancer, nor does this observation necessarily support aneuploidy over mutation. Indeed, there's nothing in mutation theory that predicts that there should be lots of newborns with cancer.
Regardless if cancer is due primarily to aneuploidy or mutation, on a strictly evolutionary basis we would expect cancer to be much less common in newborns and children because inherited susceptibility to diseases that kill before an organism reaches reproductive age are phenotypes that tend to be strongly selected against. On the other hand, genes that produce a delayed susceptibility to cancer such that it does not occur until after childbearing age in most patients would not be as strongly selected against. If those genes' effects are neutral (most common) or even somewhat advantageous in some environments (there is speculation that this is the case for BRCA1, a breast cancer susceptibility gene), then the gene remains at an equilibrium in the population. Consequently, arguing that "babies don't get cancer" (which is, in essence, what Duesberg is saying, using colon cancer as an example) is at best irrelevant to the debate over whether mutation or aneuploidy is the main cause of cancer.
In any case, infants do get cancer, although it is much less common than in the elderly, the two most common being neuroblastoma and leukemias. In fact, among children, the peak incidence of cancer is age 0-1, with a second peak around age 3. Incidence then falls until age 10, after which it starts to increase again.
Come to think of it, this argues against Duesberg, too; cancer incidence starts out higher in infants, then decreases to its lowest point in the lifespan at around age 10 (less than half the rate of infants 0-1), then starts the flat part of its sigmoidal increase to the high levels that we see in the elderly. Duesberg didn't look at the age-specific cancer incidence data finely enough. How does his aneuploidy hypothesis explain the fact that childhood cancer starts out at its highest in infants, then decreases for 10 years, then begins to increase. Mutation theory could account for it given that those infant cancers are due to congenital mutations resulting in tumors at a young age. Once children get past the age range where congenital mutations are a factor there is a very low rate of cancer, which bottoms out at around 10 years and then begins to climb as the child ages.
Can aneuploidy account for these observations better than mutation theory?
Duesberg's argument also ignores the fact that cancer is not a single disease but a collection of diseases and that the cancers that children get are generally different than the cancers that adults get. Even in children with MVA who have a very high rate of chromosome non disjunction in all their cells, the cancers they develop are still childhood cancers like rhabdomyosarcoma, leukemia and nephroblastoma. They don't, as children anyway, develop lung cancer or colon cancer or other adult cancers.
Let's be precise here.
Nowhere in the SciAm article does Duesberg claim that "babies don't get cancer".
He notes, without controversy, that cancer is typically age-dependent.
He does claim that "colon cancer is never seen in children."
In the Yang et al paper cited by Orac, the top five cancer subtypes were: leukemia in 34 infants (28.3%), retinoblastoma in 19 (15.8%), neuroblastoma in 16 (13.3%), germ cell tumor in 15 (12.5%), and brain tumor in 14 (11.7%).
But, colon cancer is nowhere mentioned.
Is there a published case report of a child with colon cancer or not?
Let's not shift goal-posts mid-argument. The editors at Scientific American plainly state that it is important to "fairly" describe the science of rivals, but I don't think it has been on this small point.
Indeed. Ky is being disingenuous by citing a "0.0%" rate of colon cancer under age 20. As the reference states, it's a rounded figure. If, for example, the true rates were 0.049%, that would round to 0.0%.
If Duesberg follows his behaviour with cases of AIDS in people not taking recreational drugs or ARV then he will simply accuse anyone under 20 with colon cancer of being a liar.
Can aneuploidy account for these observations better than mutation theory?
If you deny the existence of any observations that conflict with the aneuploidy theory of cancer then there is no problem.
His blanket claim is just plain wrong. Although it is rare in children, children can and do get colorectal cancer, and some even die of it.
See:
Kravarusic et al, Colorectal carcinoma in childhood: a retrospective multicenter study. J Pediatr Gastroenterol Nutr. 2007 Feb;44(2):209-11.
Karnak I et al. Colorectal carcinoma in children. J Pediatr Surg. 1999 Oct;34(10):1499-504.
Ameh EA and Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000 Oct-Dec;19(4):273-6.
Brown RA et al. Colorectal carcinoma in children. J Pediatr Surg. 1992 Jul;27(7):919-21.
And, really, you're being disingenuous too taking umbrage at one statement of mine and ignoring the overall argument because you think you can attack that one statement. Duesberg really does seem to be claiming that children don't get cancer. As was pointed out, his mention of the mice with oncogenes being born with normal phenotypes and not with cancer strongly implies that that was part of his argument. He used colorectal cancer as an example (and, I suspect, chose it because the sequence of genetic mutations leading to colorectal cancer have been fairly well characterized in adults). I also suspect that he chose it because it is one of those cancers that is common in the elderly and very uncommon in the young. In fact, the children who do get colorectal cancer tend to be ones with inherited mutations in colorectal cancer susceptibility genes, like mutations in APC or HNPCC, an observation that also tends to argue against Duesberg's hypothesis.
Duesberg states, "The gene mutation theory of cancer's origins, however, predicts that the disease should be quite common in newborns," and then goes on to comment about how rare cancer is in babies. (It's certainly a lot less common than in the elderly but sadly not quite "rare.") Let me repeat it for you again, since you didn't seem to get it the first two times: The reason Duesberg's argument, whether as I characterize it or as you characterize it, is so bad is because it is irrelevant to whether the mutation or aneuploidy hypothesis is correct and because the mutation hypothesis does not in any way imply or require that cancer should be common in newborns. Indeed, evolutionary theory, regardless of whether aneuploidy or mutation is the primary cause of cancer, would predict that cancer should be rare in infants and children because of negative selection against conditions that result in death before reproductive age.
Type "pediatric colorectal cancer" in the PubMed search and you get 206 hits. Barry A and "Ky" aren't interested in facts. They desperately need Duesberg to be right about everything. Because if he's wrong about colon cancer in kids, maybe he's wrong about what they really care about, like poppers and AZT supposedly causing AIDS.
Duesberg fans don't know 'APC' from an 'armored personnel carrier' and they think p24 is a cellular protein. Still they come out with their histrionic weak arguments to defend their icon. And they never concede a damn thing.
Orac like most scientists says Duesberg is right about something. There is aneuploidy in cancer. Everyone knows it and Duesberg didn't come up with it. He's jsut the only one who sticks his fingers in his ears and chants aneuploidy aneuploidy aneuploidy wherever he sees diversity of opinion.
Denialists could learn something from Orac's balanced and sensible approach.
I disagree that Duesberg might be right about aneuploidy, primarily because Duesberg wants to equate aneuploidy and chromosomal instability. I think there's both a good theoretical basis and evidence to support that chromosomal instability may be a cause or at least an early event in the development of some tumors. The description of the children with MVA and mutations in BUB1B that promote chromosome non-disjunction strongly support that idea as those kids have an apparently very high incidence of several different cancers. But where is the evidence that aneuploidy itself, as found in Down's syndrome or any of the other trisomies or monosomies found in humans, results in chromosomal instability? Duesberg argues that aneuploid embryos are not viable because they are chromosomally unstable but if that was the case then karyotyping of spontaneous abortuses should indicate that. It doesn't as far as I can tell from the literature. Reports describe trisomy 12 or trisomy 8 etc., or partial trisomy something else, not, in general, more complex karyotypes. Also, Down's syndrome children do not exhibit a higher than normal incidence of all cancers or even several cancers but only of one specific type of leukemia. In my opinion that is more consistent with the role of one or at most a few genes in the development of cancer, not the aneuploidy itself.
So whereas Duesberg argues that aneuploidy drives chromosomal/genomic instability (a notion for which I think there is little or no evidence), I think the data, such as it is, is more consistent with aneuploidy being the natural consequence of genomic instability caused by mutation or some epigenetic event.
His hypothesis is clearly testable: produce a diploid tumor, somewhere, somehow.
Hmmm...I remember that years ago, Beatrice Mintz was introducing mouse teratoma cells into mouse blastocysts, and ending up with apparently normal, cancer-free mosaic mice in which some cells were demonstrably derived from the teratoma. Based upon Duesberg's theory, this would argue that the teratoma cells somehow recovered their normal chromosome count, which sounds sort of unlikely. It would seem to fit much better with an epigenetic mechanism, at least for this rather unusual type of tumor.
I quickly skimmed over the comments, so sorry if it has already been mentioned, but:
Re: Down's: I seem to recall that the non-disjunction event can be mitotic rather than meiotic, and if it occurs early enough in development the individual will be a mosaic, with a potentially less severe phenotype. The other autosomal trisomies (Patau, Edward) are so severe that it has been hypothesised that the majority, if not all of those that survive to term are either mosaics or partial trisomies.
To all 'first-rate mediocrities' commenting on the Chromosomal Theory of Cancer here:
You guys fixate on Duesberg, while being entombed in a paradigm which demonstrably has failed. The oncongene/tumor suppressor dogma has produced no results for decades. Millions of lives and billions of dollars have been wasted on people like you pushing paper and petri dishes in thousands of labs, with no results whatsoever.
This is not an opinion, but a statement of facts. Whether it be your Rainbow Fight Against AIDS or your World War on Cancer (or the impending mad cow infected birdflu papilloma prion vaccines for underage fruitcakes), you have produced absolutely nothing, other than mountains of readily recyclable paper. (Who else but you would read them?)
Since no one forced any of you to become "scientific researchers", "doctors", or "clinical investigators", I respectfully disagree with your career choices - you would have made much better university administrators. Small stakes for small minds.
It isn't much-whined-about 'more funding' what cancer research direly needs. It's brains at long last. Instead of tens of thousands of educated paper-pushers like you, a handful of Semmelweis or Duesberg labs can produce results at a low cost.
Adele Adhominem (see above) should just take her medicine (Gleevec? why on earth would oncogenes be needed for a tyrosine kinase-inhibitor to work? molecular pathways exist in life, not because the pet theory states so and is thus mimicked by a cell who read the prevailing dogma ) then predictably die of CML or GIST in 2-5 years, while trying to figure out whether it is cancer stem cells, or progenitors or G0 that kill her in the end - or another few million possibilities in that genetic forest for the trees. All of which combinations, of course, are well contained in the chromosomal theory of cancer, but no matter. And to Orac: Warburg's cancer hypothesis? Sure, Dr. Atkins also had some ideas about it, so did my granny. Oh, and have you read any scientific researcher ever promoting someone else's theories? Please share them with me. Perhaps I misunderstood the reason behind the scientific method and the human urge to be published and reviewed. I mean, sorry, did I upset anyone or hurt anyone's feelings for not including every pet theory in the cancer field in my article? Granny, you there?
Sanderson's statement is correct. Clinical or societal benefits of the "oncogene" theory of cancer have to date been zero. He also writes "the objective is to cure cancer, and to cure something, one has to properly determine its cause. Since aneuploidy, apparently, is found in every solid tumor, it certainly must be explained -- not explained away."
No one 'invented' aneuploidy so he could get famous. Duesberg and many properly-attributed other researchers have for decades noticed aneuploidy in cancer, and consequently decided to take a closer look. Unlike you, the prevailing pet-theorists, constantly attempting to square the circle. Sorry, it does not work. Life won't imitate your theory just because it sounds good (and awards prizes) to you.
No 'improvements' of the prevailing dogma have produced any results. Nobel Prizes awarded for agreeing with prevailing views and fads are tragic. Varmus? Baltimore? Bishop? Gajdusek? (Is Weinberg next because he published a colorful new book?) In the history of science, they aint even a footnote.
Talk is cheap. Time and again, repeating those same, tired gene-theory lines, you guys have failed. Phlogistoncogenes may look pretty in your colorful, tumor-suppressed textbooks, but they neither explain nor predict cancer.
"In sum, the review of too many by too many achieves but one result with certainty: regression to the mean." - Duesberg
I'm not sure I'd go as far as a 'scientific death sentence'. But I do think that to the extent 'Peter Duesberg, Berkeley Professor' is used as an argument from authority that the work is reputable, 'Peter Duesberg, HIV crank' is a permissible counter.
It would be lovely if we could evaluate all work on its merits, as Orac has done so thoroughly here, but laypeople and even non specialist scientists don't have the time or the knowledge to do that. Some measure of 'argument from authority' is useful. It just just shouldn't be the last word.
Could we imagine an experiment, on "precancerous cells" where we could observe the very moment were the cell(s) become cancerous, and then count chromosomes to see if there are non-aneuploic cancerous cells in there ?
My guess is that it would be near-impossible to do. One spots a cancerous cell when it duplicates out of control, and by the time you can spot a mass of cells (a tumor), most of the cells would be aneuploid already. Only luck could get you a cancerous cell with its full array of chromosomes, and then, you would have a hard time to prove that it is really a cancerous cell, not a normal cell lost in a sea of aneuploic cancerous cells...
Quite a conundrum...
hi everyone.. i'm just about to graduate with my PhD in molecular genetics..
re trrll's comment on the teratoma issue...
the teratoma used in that study was diploid.